Progeria, een van de vele zeldzame aandoeningen bij de mens die verband houden met vroegtijdige veroudering. De twee belangrijkste soorten progeria zijn het Hutchinson-Gilford progeria-syndroom (HGPS), dat in de vroege kinderjaren begint, en het Werner-syndroom (volwassen progeria), dat op latere leeftijd optreedt. Een derde aandoening, het Hallerman-Streiff-François-syndroom, wordt gekenmerkt door de aanwezigheid van progeria in combinatie met dwerggroei en andere kenmerken van abnormale groei. Progeria is uiterst zeldzaam; de wereldwijde incidentie van het Hutchinson-Gilford progeria-syndroom is bijvoorbeeld ongeveer één op de vier tot acht miljoen geboorten.
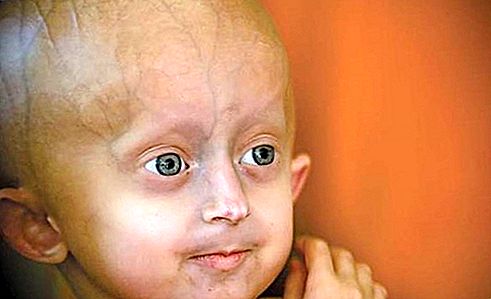
Tekenen van het Hutchinson-Gilford-progeria-syndroom verschijnen rond de leeftijd van één, na een duidelijk normale kindertijd. Getroffen personen overschrijden zelden de grootte van een normale 5-jarige, hoewel ze tegen de tijd dat ze 10 zijn het fysieke uiterlijk hebben van 60-jarige volwassenen. Veel van de oppervlakkige aspecten van veroudering, zoals kaalheid, dunner worden van de huid, prominentie van bloedvaten van de hoofdhuid en vaatziekten optreden. Geslachtsorganen blijven klein en onderontwikkeld. Een paar individuen met progeria zijn verstandelijk gehandicapt, maar de meesten hebben een normale intelligentie en kunnen zelfs vroegrijp zijn. Op de leeftijd van 10 jaar zijn uitgebreide arteriosclerose en hartaandoeningen ontstaan en sterven de meeste patiënten voordat ze de 30 bereiken; de mediane leeftijd van overlijden is 13. De aandoening is niet geërfd. Het is eerder het gevolg van de spontane mutatie van een gen dat bekend staat als LMNA (lamin A / C). In elke cel is slechts één kopie van het gemuteerde gen nodig om de ziekte te veroorzaken; daarom is het een autosomaal dominante aandoening.

Het Werner-syndroom treedt meestal op na de puberteit, met zichtbare tekenen van veroudering die het duidelijkst worden na de leeftijd van 20 jaar. De ouderdomsveranderingen zijn zodanig dat getroffen personen er ongeveer 30 jaar ouder uitzien dan hun chronologische leeftijd. Individuen bereiken mogelijk niet hun verwachte volwassen lengte, omdat de groeispurt van de adolescentie kan worden afgezwakt. Patiënten met het Werner-syndroom zijn geslachtsrijp, maar de secundaire geslachtskenmerken zijn slecht ontwikkeld. Oppervlakkige tekenen van veroudering zijn voortijdige kaalheid of vergrijzing van haar, verlies van tanden en gehoor, staar, acute artritische episodes, huidzweren en osteoporose (verlies van botweefsel). Er is een verhoogde neiging om hartaandoeningen, diabetes mellitus en kanker te ontwikkelen, en de gemiddelde levensduur is 47 jaar. Werner-syndroom wordt veroorzaakt door mutaties in het gen WRN (Werner-syndroom, RecQ helicase-achtig). Dit type progeria is erfelijk en wordt overgedragen als een recessieve eigenschap (er zijn twee exemplaren van het gemuteerde gen nodig, één van elke ouder, om de ziekte te veroorzaken).
Aanvankelijk werd progeria bestudeerd als een model van normale veroudering, maar omdat niet alle organen worden aangetast, wordt dit niet langer geschikt geacht. Er zijn bijvoorbeeld geen aanwijzingen voor seniliteit of veroudering in het centrale zenuwstelsel. Er is enig experimenteel bewijs dat suggereert dat behandeling met een medicijn dat bekend staat als n-acetylcysteïne het verouderingsproces dat gepaard gaat met het Hutchinson-Gilford progeria-syndroom zou kunnen vertragen.












